Axilatis
Elite

Bonjour !
Je vous expose donc mon petit projet

Je dispose en classe d'une dizaine de PC fixes strictement identiques.
Ils possèdent tous une carte graphique nVidia compatible CUDA.
(Il me semble que ce sont des 8800GTS mais je confirme au plus vite).
Mon but est de faire un petit cluster dans un but bioinformatique : alignement de protéines
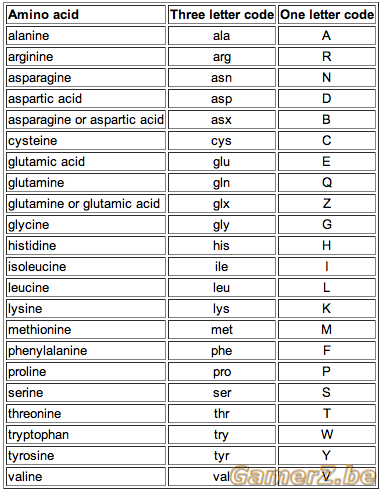
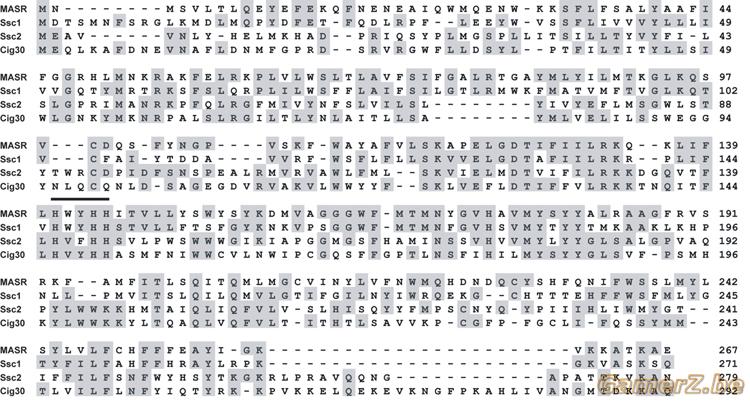
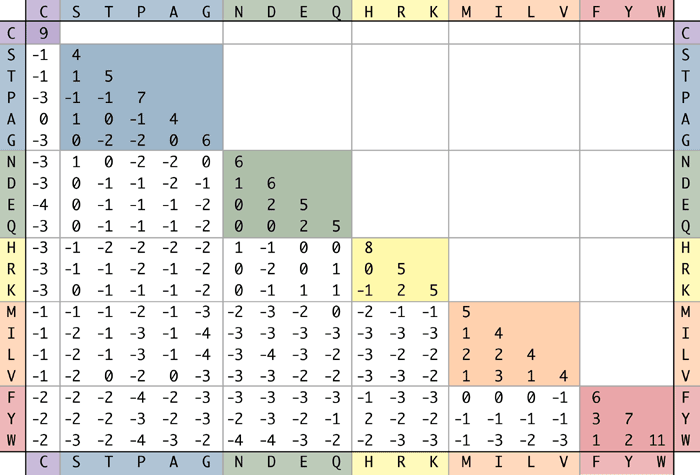
Ce qu'est un alignement de séquence (protéine ou ADN, le concept est identique).
Ce qu'est un alignement de séquence (protéine ou ADN, le concept est identique).
- 1 PC = serveur SOAP qui génère les combinaisons et envoit les séquences à aligner
- autres PCs = clients SOAP qui testent les données envoyées et retournent le score d'alignement.
- les OS utilisés : 1 client en Ubuntu (UNIX), le serveur et les autres clients en Gentoo (UNIX) (ps : c'est imposé, petit "défi" j'imagine...)



L'échange de données Clien <=> Serveur se fera par SOAP.
1 Serveur SOAP.
9 ou 10 clients SOAP (je ne sais plus exactement) qui executeront la tâche, il y aura certainement un petit script python à gauche ou à droite et forcément le logiciel/algorithme qui fera l'alignement et retournera le résultat, pour ensuite redemander de nouvelles données à aligner.
Bref je me tourne vers vous pour écouter vos conseils, vos avis, des idées sur la démarche à suivre.
Comme vous vous en doutez, ceci est un projet d'étude qui sera évalué. Je ne vous demande certainement pas de faire le travail à ma place, mais j'aimerais quelques pistes.
Peut-être se cache parmi vous un averti de CUDA qui pourrait m'aiguiller du moins pour le début??
Comme vous vous en doutez, ceci est un projet d'étude qui sera évalué. Je ne vous demande certainement pas de faire le travail à ma place, mais j'aimerais quelques pistes.
Peut-être se cache parmi vous un averti de CUDA qui pourrait m'aiguiller du moins pour le début??
Pour les autres : a dit:
D'avance, merci pour tout commentaire constructif !! :-D
~Axilatis~